
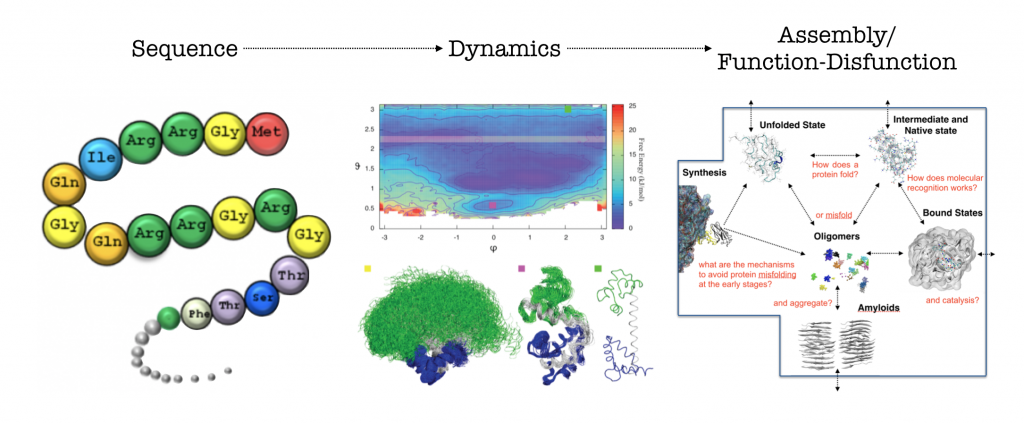
In the Computational Structural Biology lab we combine computer simulations and experiments to study biomolecules structure and dynamics. We use the resulting information to rationalise biochemical processes as well as design proteins variant and molecules to modulate them.
Active Research Grants
Computational modeling of ligand effect on human olfactory receptor 51E2 (Linea 2 – intramural funding)
In this project, our aim is to study the human olfactory receptor 51E2, which is a known target for prostate cancer. Our first step will be to model it in both its active and inactive states, based on experimental structures (where available) and de novo structures obtained using state-of-the-art AI algorithms. We will refine these models through molecular dynamics simulations. Using the relaxed/refined structures, our goal is to perform docking of experimentally known ligands, and eventually apply quantitative approaches (such as enhanced sampling) to characterize and rationalize protein-ligand interactions at the atomistic level.
Trodusquemine as a novel drug against TDP-43-associated proteinopathies: structural, biological and computational investigation – RESISTANCE (PRIN 2022 PNRR – P20225ZPYH)
This research project investigates TAR DNA-binding protein 43 (TDP-43) proteinopathies, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), which are neurodegenerative conditions characterized by a common histopathology. This pathology involves the mislocalization of TDP-43 from the nucleus to the cytosol in neurons and glia, leading to distinct inclusions that can be either ordered or amorphous aggregates. A significant focus of the project is on exploring the potential of aminosterols, particularly trodusquemine (TRO), in modulating protein aggregation and reducing toxicity. These compounds have shown promise in Parkinson’s Disease (PD) research and are undergoing clinical trials. The project aims to understand how TRO influences the accumulation and toxicity of TDP-43 in neuronal cells and a C. elegans model. It will also elucidate the protective mechanisms of TRO against TDP-43 proteinopathy both in vitro and in cultured cells, including the investigation of the inhibition of protein tyrosine phosphatase 1B as a factor in TRO’s neuroprotective effects. Thanks to the EU/MUR funding program Next Generation EU/PRIN 2022 PNRR and in collaboration with Roberta Cascella at University of Florence and Martina Banchelli at the IFAC-CNR in Florence we aim in providing new insights into therapeutic opportunities for ALS, FTD, and other TDP-43 associated proteinopathies, leveraging the diverse expertise of the research team.
Past Research Grants
Structural and functional characterization of hERG potassium channels’ enhancers (Telethon GGP19134)
The goal of this research is to test the hypothesis that small molecules that enhance the rapid component of the delayed rectifier current IKr in cardiac cells are able to rescue the phenotype in three severe variants of Long QT Syndrome (LQTS) caused by loss of function mutations in the HERG gene that encodes for the KV11.1 potassium channels that conducts IKr current (LQT2). We will apply state of the art technology to understand the relationship between structure and function and identify the binding pose of the compounds exploiting the recently resolved cryo-EM structure of the HERG channel in the open state. We will model the structure of the closed state of the channel and provide insight to our collaborators on how to improve the design of the compounds. Thanks to Telethon foundation and in collaboration with Silvia Priori at Maugeri and Giovanni Lentini at the University of Bari we will be able to perform a through assessment of a selection of the most powerful molecules previously reported in the literature and of novel molecules developed within this project.
Protein Folding, Misfolding, Aggregation and Diseases (Transition Grant – Horizon 2020)
How protein fold and how they fail are key questions to shed light on protein functional and dysfunctional behaviours. We investigate the processes at play in protein folding as well as in aggregation phenomena. We are interested in the determinants of aberrant aggregation of folded proteins to understand how such process can be modulated and regulated. We are currently studying systemic amyloidosis from beta-2-microglobulin and light chain antibodies, we are also involved in studies of protein aggregation of disordered protein in Alzheimer’s, Parkinson’s and type-2 diabetes. In particular we combine multiple simulation and experimental approaches to try to study all the relevant phases of protein aggregation: accurate equilibrium simulations combined with structural biology techniques (NMR, SAXS, X-Ray) to understand the properties of the native state, coarse-grain simulation and kinetics aggregation experiments to understand the aggregation process and cryo-EM to study the structure of the resulting fibril structures.
Joint artificial intelligence and protein structure modelling to guide large-scale screenings for anti-SARS-Cov2 neutralizing antibodies (CoroNAId)
We are part of a network lead by Stefano Casola at IFOM and in collaboration with Raffale Badolato (UNIBS/Spedali Civili di Brescia), Giulia Marchetti (UNIMI/ASST santi Carlo e Paolo) and Maria Rodriguez Martinez (IBM) were will provide a structural and dynamical understanding for the activity of large pools of antibodies heavy chain obtained from COVID-19 patients.


