New Semiclassical Theories
New Semiclassical Theories
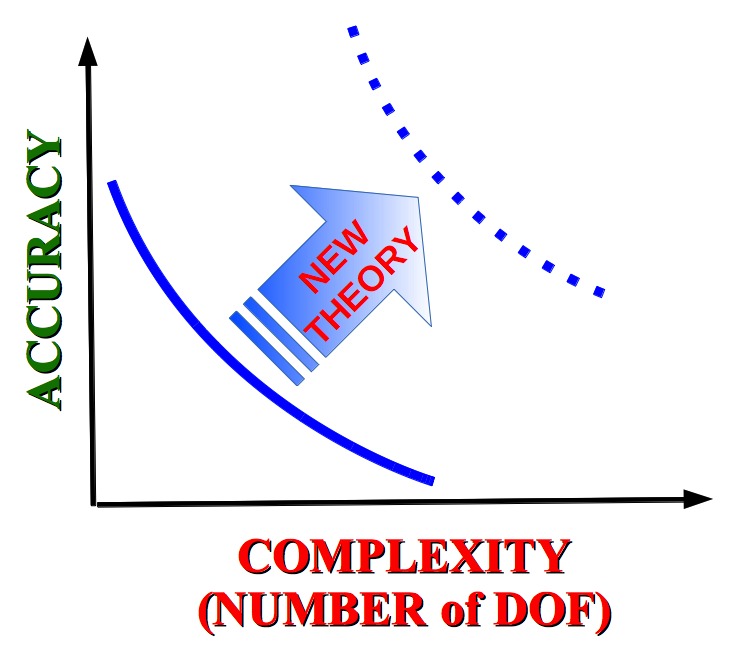
We develop new semiclassical methods for the quantum simulation of complex molecular system. The challenge is to improve simulations accuracy by adding quantum mechanical effects to molecular system with many degrees of freedom at a comparable computational cost of molecular dynamics. We further develop the semiclassical initial value representation SC-IVR invented by W. H. Miller. Theories are implemented on conventional (CPU) and innovative (GPU) computational architectures.
Suggested articles
- M. Ceotto, Y. Zhuang and W.L. Hase, 'Accelerated direct semiclassical molecular dynamics using a compact finite difference Hessian scheme', J. Chem. Phys. 138, 054116 (2013)
- M. Ceotto, 'Vibration-assisted tunneling: a semiclassical instanton approach', Mol. Phy. 110 (9-19), Special Issue 547-559 (2011)
- M. Ceotto, G.F. Tantardini, A. Aspuru-Guzik, 'Fighting the curse of dimensionality in first-principles semiclassical calculations: Non-local reference states for large number of dimensions', J. Chem. Phys. 135 (21), 214108 (2011)
- M. Ceotto, S. Atahan, G. F. Tantardini, and A. Aspuru-Guzik, 'Multiple coherent states for first-principles semiclassical initial values representation molecular dynamics', J. Chem. Phys. 130, 234113 (2009)
- M. Ceotto, S. Yang, W. H. Miller 'Quantum reaction rate from higher derivatives of the thermal flux-flux autocorrelation function at time zero', J. Chem. Phys. 122, 044109 (2005)
- M. Ceotto, W. H. Miller. 'Test of the quantum instanton approximation for thermal rate constants for some collinear reactions', J. Chem. Phys. 120, 6356 (2004)
- W. H. Miller, Y. Zhao, M. Ceotto, S. Yang. 'Quantum instanton approximation for thermal rate constants of chemical reactions', J. Chem. Phys. 119, 1329 (2003)
- S. Mandra, J. Schrier, M. Ceotto, 'Helium Isotope Enrichment by Resonant Tunneling Through Nanoporous Graphene Bilayers', J. Phys. Chem. A, 118 (33), 6457-6465 (2014)
- S. Mandra, S. Valleau, and M. Ceotto, 'Deep Nuclear Resonant Tunneling Thermal Rate Constant Calculations', Int. J. of Quantum Chemistry, 113 (12), 1722-1734 (2013)
- Y. Zhuang, M. R. Siebert, W.L. Hase, K.G. Kay, M. Ceotto, 'Evaluating the Accuracy of Hessian Approximations for Direct Dynamics Simulations', J. Chem. Theory and Computation, 9 (1), 54-64 (2013)

ab initio semiclassical molecular dynamics for complex systems
ab initio semiclassical molecular dynamics for complex systems
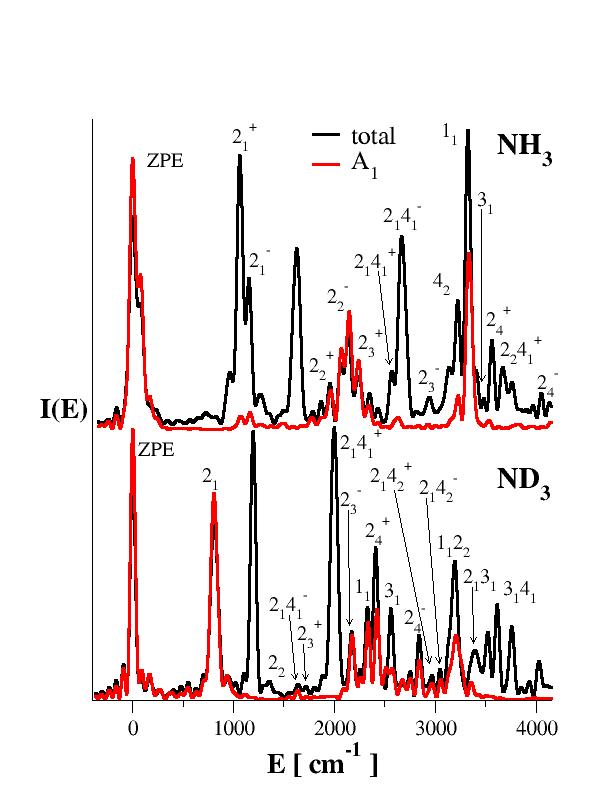
We are performing semiclassical dynamics simulation using ab initio molecular dynamics, i.e. without a pre-computed potential energy surface. Our methods add quantum mechanical effects to classical molecular dynamics simulations at a comparable computational cost of standard molecular dynamics simulations. We are focused on the calculation of nuclear molecular motion spectra (FTIR, Raman, UV-vis, CARS, Pump-probe, VCD, UPS, etc.). Our semiclassical methods can single out each spectroscopic peak. This allows a better spectroscopic and, ultimately, physical interpretation of complex molecular systems.
Suggested articles
- R. Conte, A. Aspuru-Guzik, M. Ceotto, 'Reproducing Deep Tunneling Splittings, Resonances, and Quantum Frequencies in Vibrational Spectra From a Handful of Direct Ab Initio Semiclassical Trajectories', J. Phys. Chem. Lett., 4, 3407−3412 (2013)
- M. Ceotto, S. Valleau, G.F. Tantardini, A. Aspuru-Guzik, 'First principles semiclassical calculations of vibrational eigenfunctions', J. Chem. Phys. 134 (23), 234103 (2011)
- M. Ceotto, G.F. Tantardini, A. Aspuru-Guzik, 'Fighting the curse of dimensionality in first-principles semiclassical calculations: Non-local reference states for large number of dimensions', J. Chem. Phys. 135 (21), 214108 (2011)
- M. Ceotto, D. Dell'Angelo, G.F. Tantardini, 'Multiple coherent states semiclassical initial value representation spectra calculations of lateral interactions for CO on Cu(100)', J. Chem. Phys. 133 (5), 054701 (2010)
- M. Ceotto, S. Atahan, G. F. Tantardini, and A. Aspuru-Guzik, 'Multiple coherent states for first-principles semiclassical initial values representation molecular dynamics', J. Chem. Phys. 130, 234113 (2009)
- M. Ceotto, S. Atahan, S. Shim, G. F. Tantardini, and A. Aspuru-Guzik, 'First-principles semiclassical initial value representation molecular dynamics', Phys. Chem. Chem. Phys. 11, 3861 (2009)
- M. Ceotto, G. F. Tantardini, S. Atahan, A. Aspuru-Guzik, 'First-principles implementation of semiclassical initial value representation molecular dynamics', Multidimensional Quantum Mechanics with Trajectories, pg.s-8-16, Ed. D. Shalashilin and M. Miranda (CCP6, University of Leeds, 2008) ISBN 978-0-9545289-8-0
- M. Ceotto, G. S. Ayton, G. A. Voth, 'Accelerated Superposition State Molecular Dynamics for Condensed Phase Systems', J. Chem. Theory Comput. 4, 560 (2008)
- Y. Zhuang, M. R. Siebert, W.L. Hase, K.G. Kay, M. Ceotto, 'Evaluating the Accuracy of Hessian Approximations for Direct Dynamics Simulations', J. Chem. Theory and Computation, 9 (1), 54-64 (2013)
- M. Ceotto, Y. Zhuang and W.L. Hase, 'Accelerated direct semiclassical molecular dynamics using a compact finite difference Hessian scheme', J. Chem. Phys. 138, 054116 (2013)
Semiclassical and quantum transition state theories for thermal rate constant calculations
Semiclassical and quantum transition state theories for thermal rate constant calculations
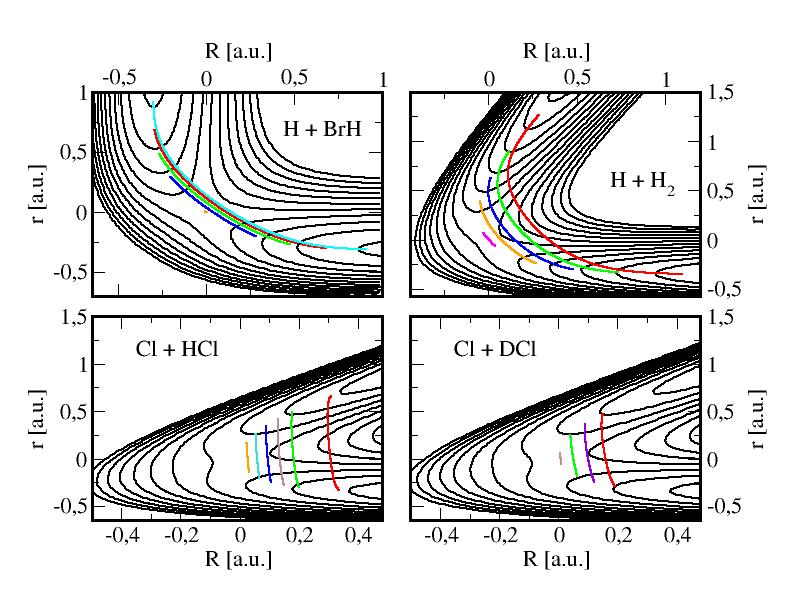
We calculate thermal rate constants by adding quantum mechanical effects, such as tunneling. We develop semiclassical Miller's theories, such as instanton and WKB based methods. Our implementation of the Monte Carlo based MultiWell suite of codes allows us to deal quantum mechanically and to understand the role of tunneling in complex reactive system.
Suggested articles

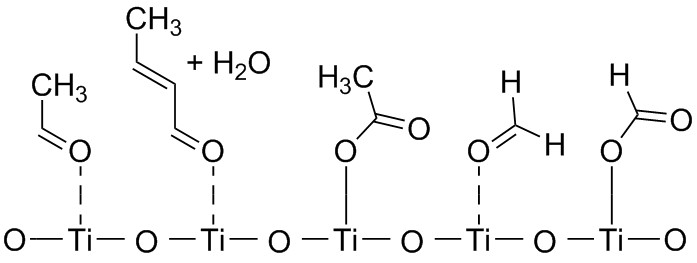
Quantum chemistry and semiclassical dynamics for titania-based environmental remediation
Quantum chemistry and semiclassical dynamics for titania-based environmental remediation
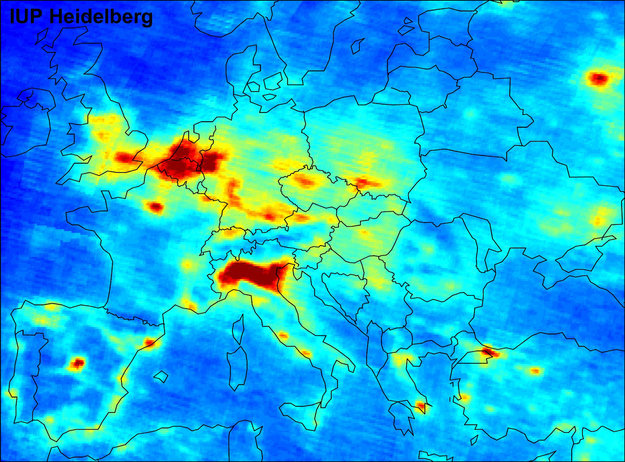
We live in one of the most polluted European city and air pollution is a major treat for many EU urban areas. The goal of this research is to explain by quantum chemistry calculations and semiclassical molecular dynamics simulations how pollutants can be transformed into carbon dioxide and water (mineralization) by the use of sunlight. Simulations are compared with electrochemical and spectroscopic partners.
Suggested articles
- M. Ceotto, L. Lo Presti, G. Cappelletti, D. Meroni, F. Spadavecchia, R. Zecca, M. Leoni, P. Scardi, S. Ardizzone, 'About the Nitrogen Location in Nanocrystalline N-Doped TiO2: Combined DFT and EXAFS Approach', J. Chem. Phys. C 116 (2), 1764-1771, (2012)
- C. Marchiori, G. Di Liberto, G. Soliveri, L. Loconte, L. Lo Presti, D. Meroni, M. Ceotto, C. Oliva, S. Cappelli, G. Cappelletti, C. Aieta, S. Ardizzone, 'Unraveling the Cooperative Mechanism of Visible-light Absorption in Bulk N, Nb Codoped TiO2 Powders of Nanomaterials', J. Phys. Chem C, 118, 24152-24164 (2014)
- L. Lo Presti, M. Ceotto, F. Spadavecchia, G. Cappelletti, D. Meroni, R.A. Acres, and S. Ardizzone, 'Role of the Nitrogen Source in Determining Structure and Morphology of N-Doped Nanocrystalline TiO2', J. Phys. Chem C, 118, 4797-4807 (2014)
- F. Spadavecchia, S. Ardizzone, G. Cappelletti, L. Falciola, M. Ceotto, and D. Lotti, 'Investigation and optimization of photocurrent transient measurements on nano-TiO2', J. of Applied Electrochem. 43 (2), 217-225 (2013)
- F. Spadavecchia, G. Cappelletti, S. Ardizzone, M. Ceotto, M. S. Azzola, L. Lo Presti, G. Cerrato, L. Falciola, 'Role of Pr on the Semiconductor Properties of Nanotitania. An Experimental and First-Principles Investigation', J. Phys. Chem C 116 (43), 23083-23093 (2012)
- D. Meroni, S. Ardizzone, G. Cappelletti, M. Ceotto, M. Ratti, R. Annunziata, M. Benaglia, L. Raimondi,'Interplay between Chemistry and Texture in Hydrophobic TiO2 Hybrids', J. Chem. Phys. C 115 (38), 18649-18658 (2011)
- . Spadavecchia, G. Cappelletti, S.Ardizzone, M. Ceotto, L. Falciola, 'Electronic Structure of Pure and N-Doped TiO2 Nanocrystals by Electrochemical Experiments and First Principles Calculations', J. Phys. Chem. C 155 (14), 6381-6391 (2011)
- D. Meroni, S. Ardizzone, G. Cappelletti, C. Oliva, M. Ceotto, D. Poelman, H. Poelman, 'Photocatalytic removal of ethanol and acetaldehyde by N-promoted TiO2 films: The role of the different nitrogen sources', Catalysis Today 161 (1), 169-174 (2011)
Quantum chemistry and semiclassical dynamics for the development of super-hydrophobic coatings
Quantum chemistry and semiclassical dynamics for the development of super-hydrophobic coatings
We simulate the development of new super-hydrophobic coatings for outdoor cultural heritage protection. We mimic nature by arranging in a periodic fashion the adsorption of hydrophobic molecules on titania films. The simulated arrangements are tested by experimental partners.
Suggested articles
- G. Cappelletti, S. Ardizzone, D. Meroni, G. Soliveri, M. Ceotto, C. Biaggi, M. Benaglia, L. Raimondi, 'Wettability of bare and fluorinated silanes: A combined approach based on surface free energy evaluations and dipole moment calculations', J. of Colloid and Interface Science 389, 284-291 (2013)
