Computational Structural Biology Lab
In the Computational Structural Biology lab we use computer simulations to study bio-molecular processes. We apply and develop integrative structural and dynamical biology methods where we combine experimental data, bio-informatics data and physicochemical knowledge to provide multi-resolution models of biomolecules in motion.
We try to understand how the conformational dynamics of biological molecules can modulate biological processes. We study the role played by conformational disorder in protein function and disfunction; the mechanisms underlying conformational changes and their role in signalling processes; the mechanisms of protein folding and misfolding and their link with protein degradation and aggregation and the mechanisms of protein-NA recognition and interaction. By using computer modelling we aim at finding general rules for these mechanisms and use them to design and propose experiments to modulate specific biological processes.

Selected Publications
- Jussupow A., Messias A.C., Stehle R., Geerlof A., Solbak S.M.O., Paissoni C., Bach A., Sattler M., and Camilloni C. (2020) The dynamics of linear polyubiquitin. SCI. ADV. 6: eabc3786
- Paissoni C., Jussupow A., and Camilloni C. (2020) Determination of Protein Structural Ensembles by Hybrid-Resolution SAXS Restrained Molecular Dynamics. J. CHEM. THEORY COMPUT. 16: 2825-2834
- Paissoni C., Jussupow A., and Camilloni C. (2019) Martini bead form factors for nucleic-acids and their application in the refinement of protein/nucleic-acid complexes against SAXS data. J. APPL. CRYST. 52: 394-402
- Camilloni C. and Pietrucci F. (2018) Advanced simulation techniques for the thermodynamic and kinetic characterization of biological systems. ADV. PHYS. X 3, 1477531
- Bonomi M., Heller G.T., Camilloni C., and Vendruscolo M. (2017) Principles of protein structural ensemble determination. CURR. OPIN. STRUCT. BIOL. 42: 106-116
Current Group

Cristina Paissoni
Postdoctoral Researcher
cristina.paissoni@unimi.it

Emanuele Scalone
PhD Student
emanuele.scalone@unimi.it

Davide Erba
MS Student
Scientific Programmes
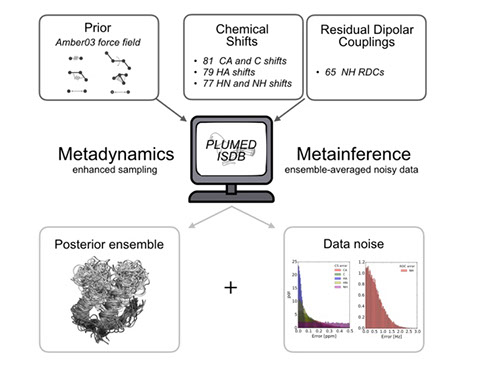
ISDB
Protein Dynamics
Protein Misfolding and Aggregation
hERG potassium channels' enhancers
ISDB
Modelling the structure and dynamics of biomolecular systems requires the integration of multiple sources of knowledge. Experiments can generally provide accurate structural information and lower resolution indication about the dynamics of a system. Molecular simulations can provide high-resolution dynamics with limitations in its accuracy. We develop theoretical and computational tools to integrate experimental information in molecular dynamics simulations thus enabling high-resolution characterization of both structure and dynamics.
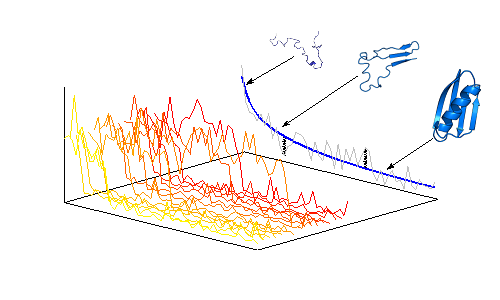
Protein Dynamics and Molecular Recognition
Proteins move, fluctuate around their native ground state and exchange among low populated excited states. Such substates can be exploited to facilitate the binding between proteins, proteins and nucleic acids or other molecules. This behaviour is particularly evident in the case of disordered proteins that by lacking a specific tertiary structure exchange continuously among a large number of weakly populated states. We use molecular modelling to characterise protein dynamics and correlate it with functional and thermodynamic features. We are currently studying the role of dynamics in the inflammatory processes mediated by linear-poly-ubiquitination, the role of dynamics in protein-RNA binding, the role of disorder in protein recognition and binding.
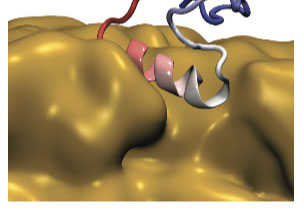
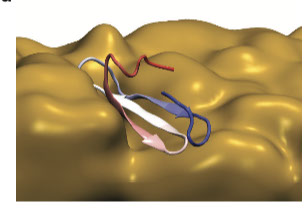
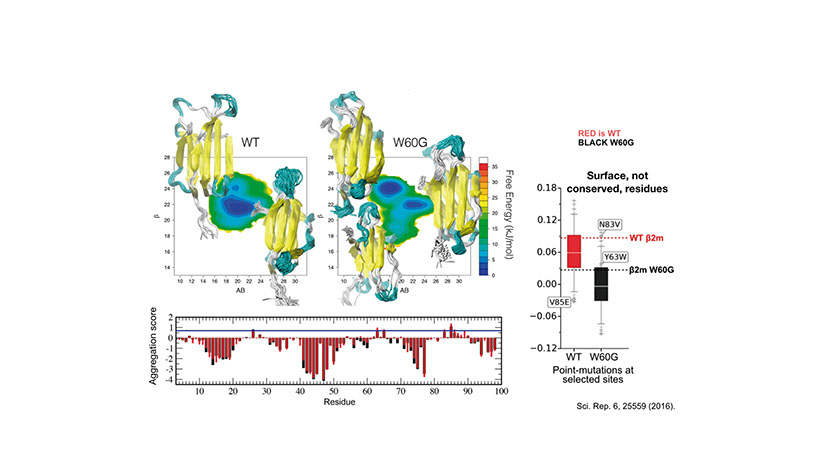
Protein Folding, Misfolding and Aggregation
How protein fold and how they fail are key questions to shed light on protein aggregation. We are interested in shedding light on the determinants of successful folding as well as of aberrant aggregation of folded and intrinsically disordered proteins. We are currently studying systemic amyloidosis from light chain antibodies, beta-2-microglobulin and type-2 diabetes. Furthermore we always try push the boundaries for the use of MD simulations in understanding protein folding.
Structural and functional characterization of hERG potassium channels' enhancers (Telethon GGP19134)
The goal of this research is to test the hypothesis that small molecules that enhance the rapid component of the delayed rectifier current IKr in cardiac cells are able to rescue the phenotype in three severe variants of Long QT Syndrome (LQTS) caused by loss of function mutations in the HERG gene that encodes for the KV11.1 potassium channels that conducts IKr current (LQT2). We will apply state of the art technology to understand the relationship between structure and function and identify the binding pose of the compounds exploiting the recently resolved cryo-EM structure of the HERG channel in the open state. We will model the structure of the closed state of the channel and provide insight to our collaborators on how to improve the design of the compounds. Thanks to Telethon foundation and in collaboration with Silvia Priori at Maugeri and Giovanni Lentini at the University of Bari we will be able to perform a through assessment of a selection of the most powerful molecules previously reported in the literature and of novel molecules developed within this project. We will therefore test six compounds in: 1) human induced pluripotent stem cells (hiPSC)-derived cardiac myocytes from carriers of different pathogenic mutations, 2) guinea-pig models of LQT2 and LQT3 and 3) in a knock-in swine model of LQT8 that we have recently developed.


The Structural Biology Group comprises members from both the DBS-UNIMI and the IBF-CNR. The content herein is not regulated by the University of Milan.