Accelerated Molecular Dynamics for Peptide Folding: Benchmarking Different Combinations of Force Fields and Explicit Solvent Models
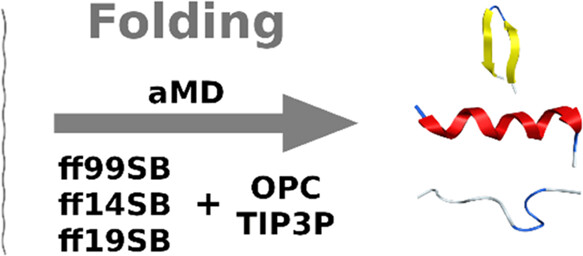
Accelerated molecular dynamics (aMD) protocols were assessed on predicting the secondary structure of eight peptides, of which two are helical, three are β-hairpins, and three are disordered. Protocols consisted of combinations of three force fields (ff99SB, ff14SB, ff19SB) and two explicit solvation models (TIP3P and OPC), and were evaluated in two independent aMD simulations, one starting from an extended conformation, the other starting from a misfolded conformation. The results of these analyses indicate that all three combinations performed well on helical peptides. As for β-hairpins, ff19SB performed well with both solvation methods, with a light preference for the TIP3P solvation model, even though performance was dependent on both peptide sequence and initial conformation. The ff19SB/OPC combination had the best performance on intrinsically disordered peptides. In general, ff14SB/TIP3P suffered the strongest helical bias.
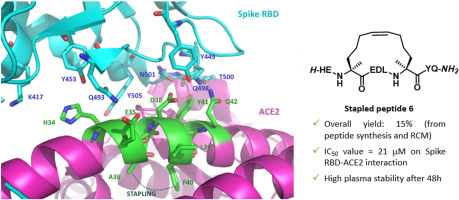




comments