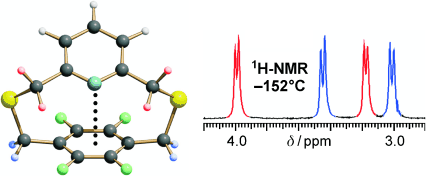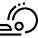

Chiral supramolecular assemblies
Aromatic tripodal receptors for (C60-IH)[5,6] fullerene
G. Dell'Anna, M. Benaglia, R. Annunziata, F. Cozzi, G. Celentano, O. Francesconi, S. Roelens, Org. Biomol. Chem., 2009, 7, 3871-3877 [Link]
Abstract: Monotopic and ditopic tripodal benzene platforms featuring aromatic and perfluoroaromatic side-arms have been synthesized, and their binding properties toward C60-fullerene have been investigated by HPLC examining retention times on a fullerene-modified silica stationary phase, using highly polar eluants (acetonitrile and acetonitrile/water). By comparison of structurally homogeneous sets of receptors, a clear trend could be found, pointing to an increased retention for ditopic derivatives, in which binding can occur on both sides of the benzene platform, over their monotopic counterparts. Among the latter, monotopic receptors containing H-substituted aromatic residues showed stronger retention than their perfluorinated analogues. This effect was ascribed to the greater availability of the π-electrons in a H-substituted aromatic ring with respect to the corresponding F-substituted counterpart in participating in a π–π interaction with the electron-poor surface of fullerene. Several NMR experiments aimed to investigate binding interactions in solution, using the much less polar solvents required by the fullerene solubility (1,1,2,2-tetrachloroethane, chloroform, toluene, and CS2), did not provide any evidence of binding interactions. We concluded that π–π interactions between fullerene and the investigated flexible tripodal receptors cannot compete with solvation in poorly polar solvents, and that the binding interactions observed by HPLC were essentially forced by the strongly polar eluant employed for the HPLC analysis.
Synthesis of some terpyridines disubstituted in positions 6 and 6' with head to tail oriented amino Acids and Dipeptides: A Simple Entry to a Reversible Inducer of Folding in Amino Acid Sequences.
R. Annunziata, M. Benaglia, A. Puglisi, F. Cozzi, L. Raimondi, Eur. J. Org. Chem,, 2008, 23, 3976-3983 [Link]
Abstract:The 2,2':6',2''-terpyridine scaffold has been identified as a conformationally discrete structural element potentially capable of inducing reversible folding in substituents, attached through suitable spacers to its 6,6″-positions, by metal complexation/decomplexation or by protonation/deprotonation. The synthesis of some terpyridine–amino acids and terpyridine–dipeptide conjugates is described. The assembly of these conjugates has been achieved by connecting NH2- and CO2H-protected glycine, alanine, and valine residues or antiparallel oriented AlaGly/GlyAla chains to the terpyridine scaffold through phenylacetylene spacers. Preliminary experiments showed that upon addition of Zn2+ to the amino acid substituted transoid terpyridine systems, folded cisoid complexes were formed. Also, bis(protonation) of the dipeptide-substituted system resulted in the formation of a folded adduct. Reversibility of the folding process was shown by Zn2+ removal with triethylamine or deprotonation with aqueous ammonia.
A molecular gate: control of the free intramolecular rotation by application of an external signal.
R. Annunziata, M. Benaglia, M. Cinquini, F. Cozzi, L. Raimondi, J.Phys. Org. Chem., 2004, 17, 749-751. [Link]
Abstract: Control of internal movement in a conformationally free pentiptycene derivative is achieved by complexation of two phenanthroline units by copper(I) cation.
See also
The Intramolecular Interaction of Thiophene and Furan with Aromatic and Fluoroaromatic Systems in some[3,3]meta(heterocyclo)paracyclophanes: a Combined Computational and NMR study.
M. Benaglia, F. Cozzi, A. Mazzanti, M. Mancinelli, Chem. Eur. J., 2010, 16, 7456-7468. [Link]
Abstract: Four thiophene- and furan-containing [3.3]meta(heterocyclo)paracyclophanes were designed and synthesized to study the intramolecular interaction between standard heteroaromatic rings and tetra-H- or tetra-F-substituted benzenes. A complete conformational analysis, carried out by DFT calculations and variable-temperature NMR techniques, showed that, despite their structural similarity, these adducts have different conformational preferences and undergo different types of isomerizations depending on the nature of the heterocycle. The thiophene-derived adducts adopted a parallel stacked arrangement of the aromatic systems in the ground-state conformations. Their isomerization pathways involved a thiophene ring-flip process passing through an edge-to-face arranged transition state in which the heterocycle is perpendicular to the benzene platform and its sulfur atom points toward the center of that ring. The threshold energy barrier to the ring-flip process was higher by 10 kJ/mol −1 in the case of the adduct featuring the perfluorinated benzene. This difference was rationalized by assuming that the ground-state conformations of the H- and F-substituted compounds have different stability. On the contrary, the furan-derived adducts were shown by calculations and NMR spectroscopy to adopt, in their ground-state conformations, a perpendicular edge-to-face disposition of the rings with the oxygen atom pointing toward the benzene platform. The adoption of this arrangement was confirmed by X-ray crystallography. In the case of these compounds, the isomerization process involved distortion of the CH2SCH2 bridges connecting the aromatic systems and the adoption of transition-state geometries for which the rings were arranged in a parallel-stacked orientation. Once again a very nice agreement was observed between the predicted and the experimentally determined geometries and pathways. In the case of the furan-containing compounds, the threshold barriers were found to be much lower in energy that those observed for the thiophene derivatives. Remarkably, they were virtually independent of the presence of fluorine atoms on the platform benzene ring.

The Intramolecular Edge-to-Face Interactions of an Aryl C-H Bond and of a Pyridine Nitrogen Lone Pair with Aromatic and Fluoroaromatic Systems in Some [3,3]Metaparacyclophanes: A Combined Computational and NMR Study.
R. Annunziata, M. Benaglia, F. Cozzi, A. Mazzanti, L. Lunazzi, Chem. Eur. J., 2009, 15, 4373-4381. [Link]
Abstract: Having the edge on aromatic - aromatic interactions: Model systems containing aromatic, fluoroaromatic, and heteroaromatic residues arranged in an edge-to-face disposition have been synthesized (see figure). Investigation of their structures and stereodynamic behavior shows a favorable interaction between an "edge" ring featuring a pyridine nitrogen lone pair and a perfluorinated “face” ring. Simple model systems based on the [3,3]metaparacyclophane skeleton have been designed to study the effect of fluorination of the “face” ring on the edge-to–face (EtF) interactions with the CAr-H bond of a phenyl ring or the nitrogen lone-pair of pyridine. Calculations established that in their more stable conformation the model systems adopt a tilted EtF disposition with the rim of the meta-substituted ring pointing towards the face of the para-substituted ring. Topomerization occurs by flipping of the meta-substituted ring, a process that involves the formation of an intermediate featuring an orthogonal EtF disposition of the arenes, which is less stable than the tilted arrangement. The energy barriers to the isomerization were determined by variable-temperature NMR spectroscopy and were well reproduced by DFT calculations. The variation in the energy barrier as a function of the substitution of the para-substituted ring could be rationalized well by a polar interpretation of the EtF interaction in the cases of model systems presenting the PyN ... π interaction but not in the cases of models featuring the CAr-H...π interaction.